
The Nitrogenase Nuance
Chemists reveal one mechanism of dihydrogen production by nitrogenase
(November 2017)
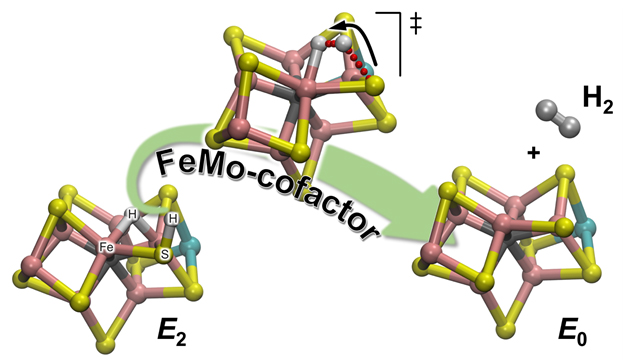
Schematic illustration of how H2 is formed from one of the reaction enzymatic intermediates (E2) to regenerate the resting state (E0) of the nitrogenase catalytic cofactor, called FeMo-cofactor. The hydrogen atom bound to one of the sulfur atoms initiate a journey that will take it to meet with the partner with which it will form the H2 molecule: one hydridic hydrogen patiently waiting, sitting between two Fe atoms. Enlarge Image.
Nitrogenase is central to life on our planet. It provides most of the nitrogen (N) that is used in proteins and nucleic acids and it's essential in creating plants, animals and other organisms, making ammonia (NH3) which is then used in the biosynthesis of amino acids and then proteins. But while nitrogenase is at work, it's also creating something else: dihydrogen (H2). Scientists have known for some time that nitrogenase makes H2 by two different processes. Now, one of these processes is clear through the work of a team of researchers who studied the mechanism for H2 production.
Nitrogenase will make H2 if run in the absence of N2. This has been viewed as a "relaxation" of the enzyme active site that is charged with iron-hydrides that are needed for the N2 reduction reaction. But what is the mechanism for how the nitrogenase active site iron-hydrides relaxed to make this H2? A team of scientists from Pacific Northwest National Laboratory, Utah State University, Northwestern University, and the University of Utah sought to understand this H2 relaxation mechanism by monitoring the effects of hydrogen (H) vs deuterium (D) on the kinetics of H2 formation when nitrogenase is attached to an electrode. The team discovered that a nearby H+ was actually the active partner in the production of H2 when it combines with the iron-hydride to make H2.
"Only one proton does everything, the hydride is just a mere spectator waiting for some action," confirmed Simone Raugei, a PNNL scientist involved in this work.
Why It Matters: This is an important step toward understanding nitrogenase and how and why the production of H2 is required for its activity. It also illustrates a strategy for mechanistic study that can be applied to other oxidoreductase enzymes and to biomimetic complexes.
Summary: To isolate the kinetics of hydrogen production, a team led by Lance Seefeldt, Brian Hoffman, Shelley Minteer, and Simone Raugei used small molecules to quickly shuttle electrons from an electrode to the catalytic half of nitrogenase. Following the proton reactions was a tricky and challenging task, said Seefeldt, so to ensure accuracy they monitored the proton "inventory" (the number of hydrogens involved in the catalytic production of H2) by performing kinetic hydrogen/deuterium kinetic measurements.
The results of the experiments suggested that only one hydrogen was actually involved in the rate-limiting step of H2 formation, and not two as the stoichiometry of the reaction indicates.
Cleared slide summarizing research conducted at Pacific Northwest National Laboratory. Download PowerPoint slide.
The team's experimental observations were upheld through computational chemistry, which provided a detailed atomistic description of the reaction mechanism. There are 2 types of hydrogens in the catalytic cofactor (called FeMo-co): one located in a bridging position where it binds to two iron atoms (a bridging hydride), and one on a nearby sulfur atom ligands (basically a protonated sulfur atom: S-H). It turns out that during H2 evolution, the latter is the active partner. The S-H bond breaks and the resulting H+ moves toward the bridging hydride and protonates it forming H2: The calculations provide an explanation of the experimental observation that only one hydrogen is "active," while the other is a mere spectator waiting for the arrival of its partner.
"The level of agreement between the observations and the computational chemistry results was remarkably high," said Raugei.
This work is a critical step toward a mechanistic understanding of the nitrogenase enzyme. There is still work to be done to understand the mechanism of the other H2 evolution process that leads to N2 binding, activation, and reduction to NH3.
Acknowledgments
Sponsors: Work by Lance C. Seefelt and Dennis R. Dean was supported by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences (BES) under awards DE-SC0010687 and DE-SC0010834. Work by Simone Raugei was also supported by the DOE, Office of Science, BES, Chemical Sciences, Geosciences, and Biosciences Division under contract DE-AC05-76RL01830/FWP66476. Work by Brian M. Hoffman was supported by the National Institutes of Health under award GM 111097. Work by Ross D. Milton was supported by a Marie Curie-Sklodowska Individual Fellowship (Global) under the European Commission's Horizon 2020 Framework (project 654836).
Research Area: Chemical Sciences
User Facility: All of the quantum chemical calculations were performed at EMSL, a DOE Office of Science user facility.
Research Team: Nimesh Khadka (Utah State University), Ross D. Milton (University of Utah), Sudipta Shaw (Utah State University), Dmitriy Lukoyanov (Northwestern University), Dennis R. Dean (Virginia Tech), Shelley D. Minteer (University of Utah), Simone Raugei (Pacific Northwest National Laboratory), Brian M. Hoffman (Northwestern University), and Lance C. Seefeldt (Utah State University)
Reference: Khadka N, RD Milton, S Shaw, D Lukoyanov, DR Dean, SD Minteer, S Raugei, BM Hoffman, and LC Seefeldt. 2017. "Mechanism of Nitrogenase H2 Formation by Metal-Hydride Protonation Probed by Mediated Electrocatalysis and H/D Isotope Effects." Journal of the American Chemical Society 139:13518-13524. DOI: 10.1021/jacs.7b07311
Highlight written by Michelle Ma and Simone Raugei